環境微生物のゲノム多様性を高解像度に検出 ―「似て非なるゲノム」から生物多様性の源泉に迫る―
環境中の微生物のゲノム情報を、培養を経ず直接的・網羅的に取得する「メタゲノム解析」の登場で、微生物の多様性をとりまく我々の理解は飛躍を遂げました。一方で、メタゲノム解析には、環境中に共存する極めて近縁な「似て非なる」ゲノムを区別することが難しいという弱点がありました。
京都大学化学研究所 岡﨑友輔 助教 (研究開始時:産業技術総合研究所 日本学術振興会特別研究員)、同大学生態学研究センター 中野伸一 教授、国立遺伝学研究所 豊田敦 特任教授、産業技術総合研究所生物プロセス研究部門 玉木秀幸 副研究部門長らの共同研究グループは、従来法では捉えられなかった環境中の細菌ゲノムにおけるわずかな変異を塩基多型・構造多型の両側面から網羅的に検出可能なメタゲノム解析法を確立し、琵琶湖に生息する細菌群集の多様性の実態を高解像度で明らかにしました。さらにその結果の解析から、ウイルス感染への抵抗性、および細菌群集の集団サイズがゲノムの多様化をつかさどる主要因であることを示しました。「似て非なる」ゲノムの比較解析から生物多様性の源泉に迫った本研究は、環境中の微生物の多様性を高解像度に捉える研究の必要性を示し、微生物の進化と生態をとりまく理解を知見と手法の両側面から新たな段階へと導く成果といえます。
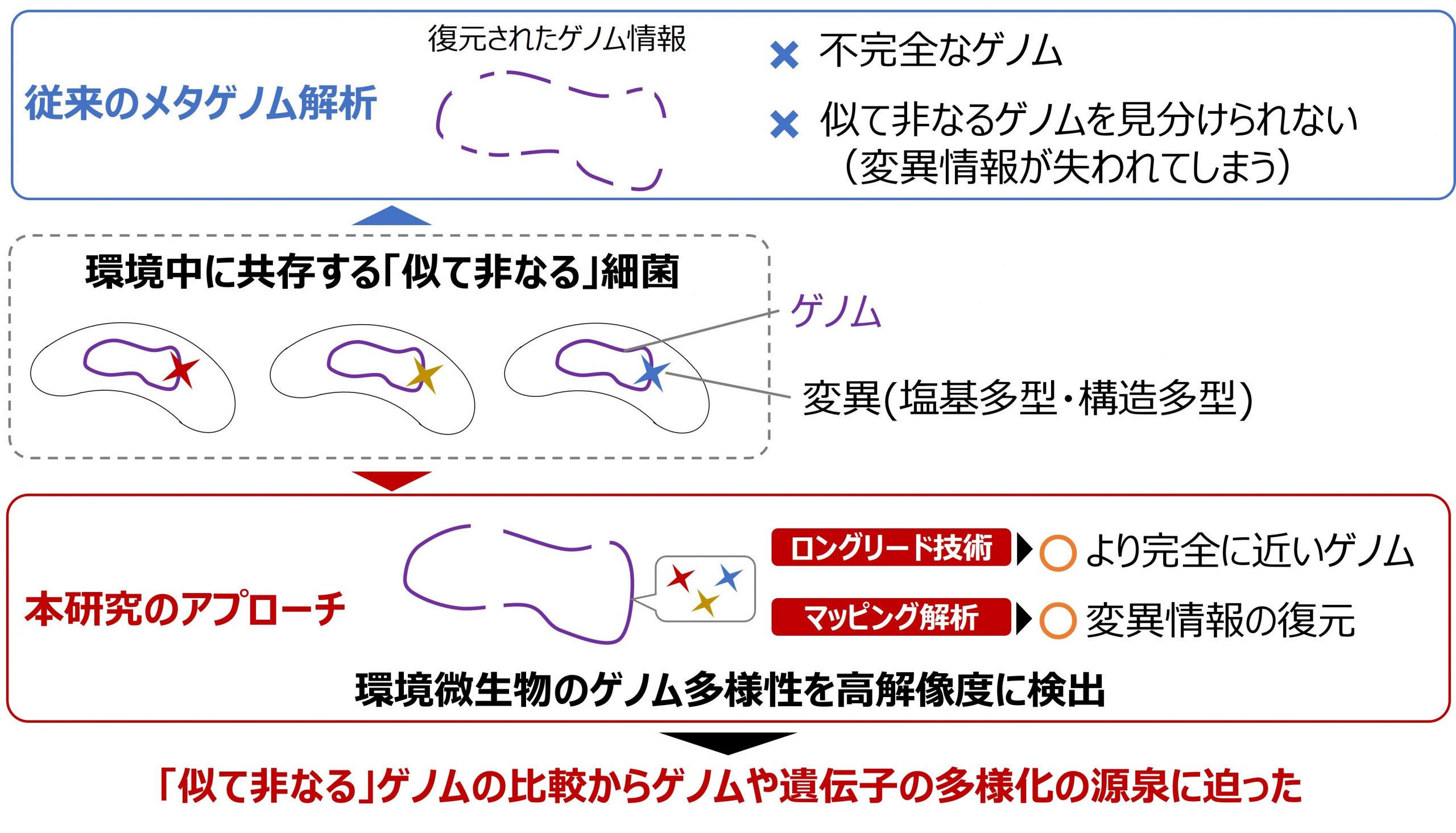
図:本研究の概要図
細菌は地球上のあらゆる環境に生息し、その数の総計は宇宙の星の数の約100万倍、10の30乗細胞とも推定されています。例えば、湖や海の水コップ1杯の中には、日本の人口に匹敵する1億を超える数の細菌が存在しています。その生き様を明らかにすることは、生態系や物質循環の基盤をなす微生物の働きを理解するうえで不可欠です。しかし、環境中の細菌の約9割が難培養微生物であり、その多様性や生態の理解には多くの技術的困難を伴います。近年のDNAシーケンス技術の急速な発展に伴って登場した「メタゲノム解析」は、環境中の微生物の全ゲノム情報を、培養を経ることなく、直接的に取得することを可能にし、この課題に対する強力な打ち手として急速な普及を見せています。今や我々が知る地球上の微生物多様性のほとんどが、メタゲノム解析によって明らかになった「培養株は存在しないがゲノム塩基配列としてその存在が知られている」系統で占められるに至っています。
一方でメタゲノム解析では、環境中の多様な微生物に由来する、純粋ではないDNAから断片配列を取得し、そこから元のゲノム配列を復元するという工程を経るため、その精度には限界があることが知られています。特に、系統的に極めて近縁な、ゲノムの塩基配列が似た複数種の微生物が共存している場合、その微小な多様性を見分けることが難しいという問題があります。簡単に言えば、「複雑なものを単純化するために、似て非なるものには目をつぶる」のがメタゲノム解析の背景にある考え方であり、環境微生物の真の多様性を明らかにするには解像度が足りないという問題がありました。
そこで本研究では、この問題を克服する高解像度なメタゲノム解析法を確立し、従来法では捉えられなかった環境中の「似て非なる」細菌ゲノム間の多様性の実態を明らかにするとともに、そのゲノム多様化の背景にある要因を明らかにすることを目的に研究を行いました。
本研究では従来のメタゲノム解析法に、ロングリードシーケンスとリードマッピング解析を組み合わせることで、極めて近縁な系統に対して、高い解像度での解析を実現しました。対象とする微生物サンプルは、京都大学生態学研究センターの調査船「はす」(写真1)を用いて、2018年5月から2019年4月の12か月の毎月、琵琶湖沖の表層(水深5 m)および深層(水深65 m)から採集しました(図1、写真2)。琵琶湖沖では水深と季節の変化に対応して様々な微生物系統が出現することが知られており、その多様性を網羅した横断的な比較解析ができるメリットがあります。得られたサンプルからDNAを抽出し、従来法(ショートリードシーケンス)にロングリードシーケンスを組み合わせたメタゲノム解析を行いました。その結果、琵琶湖に生息する主要な系統を網羅した、21の門を含む、575個の細菌および古細菌のゲノム情報が得られました。そのうち188個がゲノム断片数が10未満、さらに29個は完全長ゲノムであり、従来法では達成できなかった極めて高品質なゲノム情報の復元に成功しました。しかしこの段階では、575個のゲノムは、わずかに異なった複数のゲノムを代表する塩基配列として得られているため、「似て非なる」ゲノムを見分けるには至っていません。
そこで、得られたゲノムに対しリードマッピング解析を行い、575個のそれぞれのゲノムの中に存在する微小な多様性を検出しました。本研究の特色として、ゲノム上に存在する塩基多型と構造多型の両者を、環境微生物群集を対象に網羅的に決定した点が挙げられます。このうち構造多型はゲノムの生理・生態的特性を変化させうる重要な変異を含みますが、従来法(ショートリードシーケンス)で検出できるのは塩基多型のみであり、構造多型の検出は原理的に困難でした。本研究では、ロングリードシーケンスとマッピング解析の二つの先端的手法を相乗することで、環境中の難培養微生物のゲノム上の構造多型の全容を明らかにすることに成功しました。その結果、得られた575個のゲノムそれぞれについて多数の変異が検出され、塩基配列や遺伝子組成がわずかに異なる多数の「似て非なる」ゲノムが環境中に共存している実態を明らかにしました(図2、図3)。
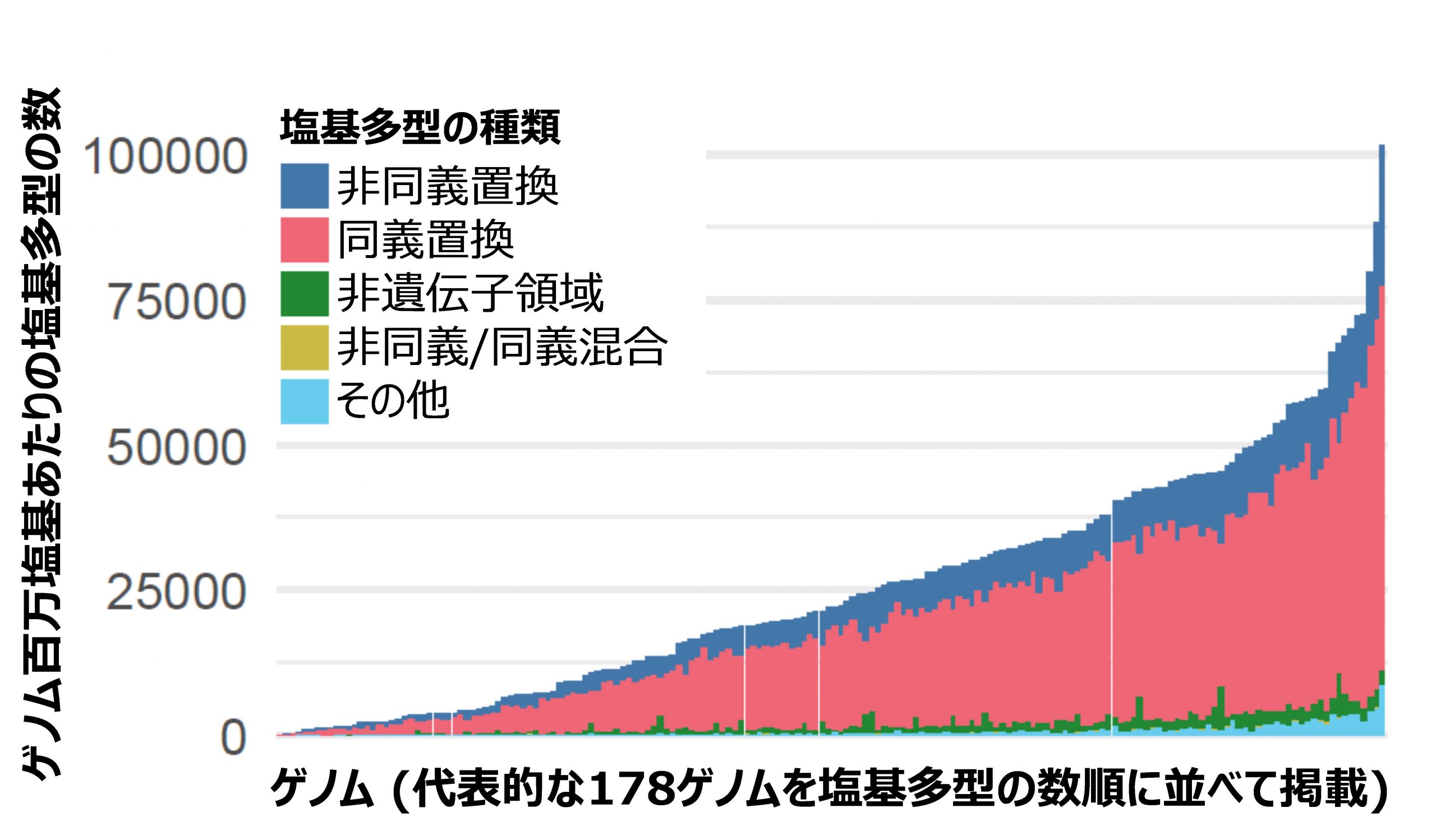
図2:各ゲノムで検出された塩基多型の数
ゲノム内に含まれる塩基多型の数はゲノム間で大きく異なり、同義置換(アミノ酸配列に変化を与えない塩基置換)が過半を占める。
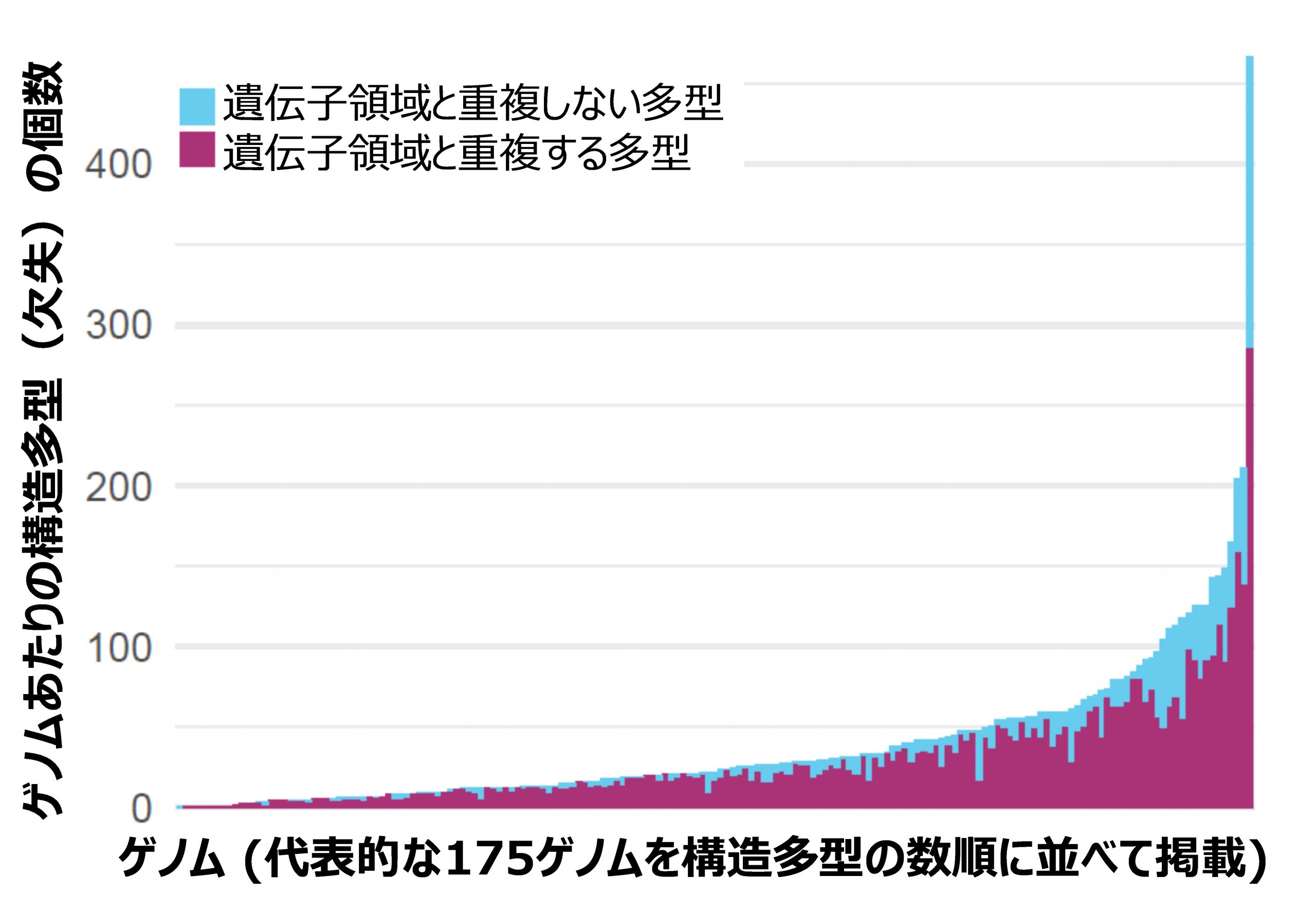
図3:各ゲノムで検出された構造多型(欠失)の数
ゲノム内に含まれる構造多型の数はゲノム間で大きく異なり、遺伝子領域と重複する多型が全体の8割以上を占める。
さらにこれらの「似て非なる」ゲノム間のわずかな違いを、系統間・遺伝子間で横断的に比較し、その多様性の背景にある要因を探りました。その結果、塩基多型、構造多型とも、ウイルス感染からの逃避に関わる遺伝子領域で起こりやすいことが明らかとなり(図4)、ウイルス感染が環境中の細菌ゲノムの多様化を駆動する主要因であることを示しました。一方で、季節・水深間の横断的な比較からは、細菌の集団サイズの減少(ボトルネック)が、ゲノム多様化の大きな制約となっている実態を明らかにしました。また逆方向からのアプローチとして、ゲノムの微小多様性の程度を知ることで、過去に集団サイズのボトルネックが存在したかどうかを推測できる可能性を示しました。
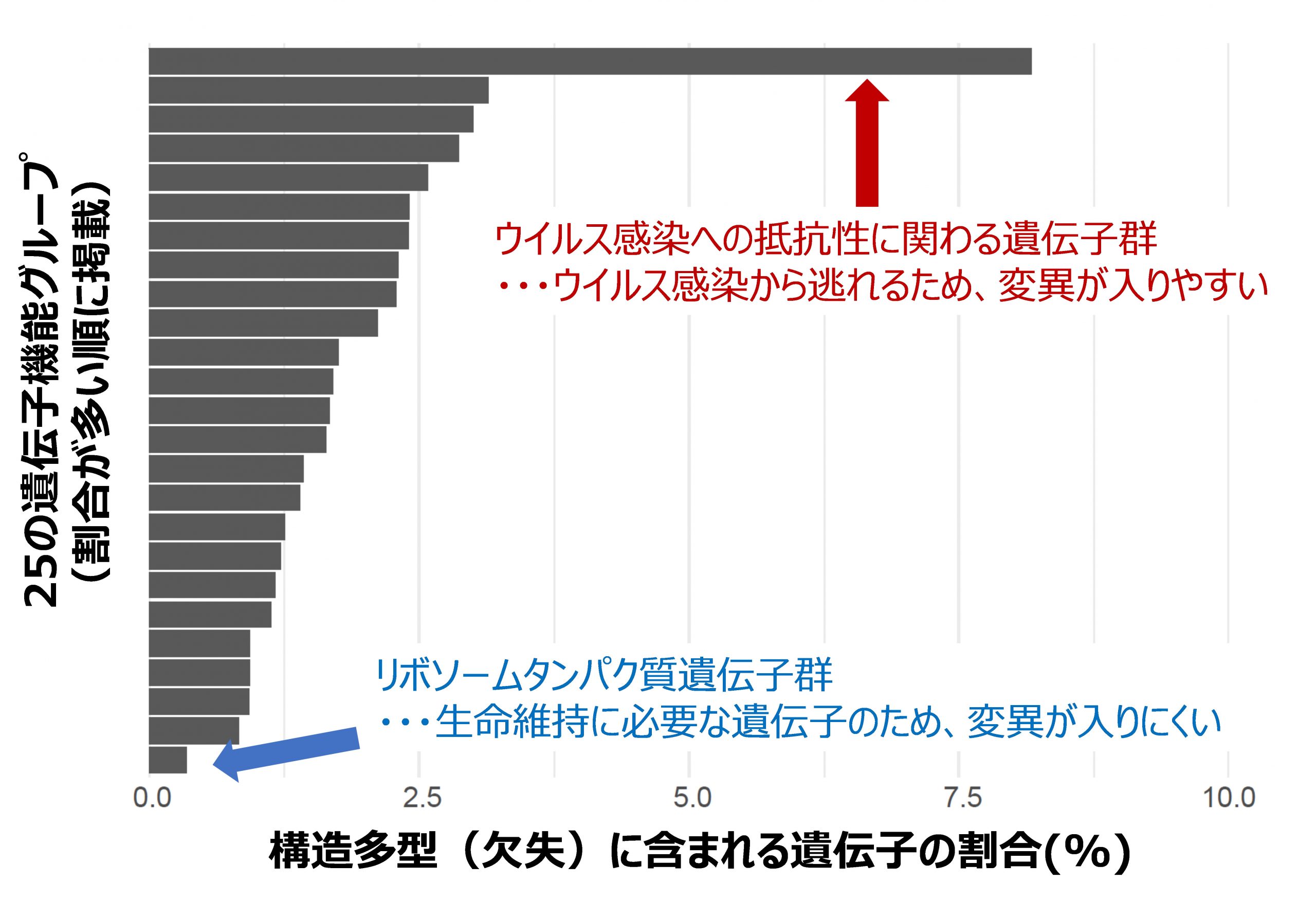
図4:遺伝子機能グループごとの構造多型(欠失)に含まれる遺伝子の割合
解析した全ゲノムの全遺伝子を主要な25の機能グループに分類して比較した。ウイルス感染への抵抗性に関わる遺伝子群で、突出して構造多型が多く見られた。
本研究では、ロングリード技術をメタゲノム解析に応用し、それを琵琶湖で時空間的に採集した微生物サンプルに適用することで、「琵琶湖産細菌ゲノムカタログ」ともいえる、高品質かつ網羅的な細菌ゲノム情報を構築しました。この情報は、今後の湖沼微生物生態学において基盤的情報として広く活用されることが期待されます。
さらにこの高品質ゲノム情報を対象に、その背景に隠されたわずかな変異を塩基多型・構造多型の両側面から網羅的に検出する手法を確立し、その多様性の実像を、従来法では届かなかった高い解像度で示しました。「似て非なる」ゲノムの比較解析から生物多様性の源泉に迫った本研究は、環境微生物のゲノム多様性を高解像度に捉える研究の必要性を示し、知見と手法の両側面からその可能性を拓く成果といえます。
今後は、琵琶湖以外の湖でも同様の研究を展開し、湖間で「似て非なる」ゲノムの多様性を比較することで、「異なる湖の微生物にはどの程度の遺伝的な差異があるのか?」「異なる湖でゲノム多様化の方向性はどの程度共通/相違しているのか?」といった問いの答えを追求していきます。それにより、本研究で得られた仮説を複数の湖で検証するとともに、それぞれの湖で起こった異なる進化の道筋を炙り出し、その背景にある微生物ゲノムの普遍的な多様化メカニズムに迫りたいと考えています。
本研究は、JSPS 科学研究費補助金「先進ゲノム支援 (PAGS)(16H06279)」、「特別研究員奨励費(18J00300)」、「基盤研究(B)(19H03302)」、「若手研究(22K15182)」、「基盤研究(A)(22H00382)」、京都大学教育研究振興財団 研究活動推進助成、京都大学生態学研究センター 共同利用・共同研究事業の支援を受けて実施されました。 また、本研究成果は、京都大学化学研究所スーパーコンピュータシステムを利用して得られたものです。
●用語解説●
メタゲノム解析:環境中の微生物群集から抽出した、複数種が混在した状態のDNAをシーケンサーで解読し、培養を経ることなく各微生物系統のゲノム情報を得る方法。難培養微生物を対象とした研究手段として微生物生態学で広く用いられている。
塩基多型:ゲノム上に存在する、DNAの塩基レベルの多型。遺伝子領域に塩基多型が起こった場合、遺伝子の機能に影響を与える場合がある。
構造多型:ゲノム上に存在する、DNA配列断片レベルの多型。挿入・欠失・逆位・重複が含まれる。遺伝子領域に構造多型が起こった場合、遺伝子の獲得・欠損・機能不全を引き起こす場合がある。
難培養微生物:培養方法が確立していない、または培養が極めて難しい微生物系統。単離培養株を用いた実験が行えないため、研究手段が限られる点が課題である。
ロングリードシーケンス:数千塩基を超える長いDNA配列を解読できるシーケンス技術。一般的に、Oxford Nanopore社(本研究で使用)およびPacific Biosciences (PacBio)社のシーケンサーを指す。数百塩基の長さまでしか解読できない従来型のDNAシーケンサー(ショートリードシーケンサー)と比較すると、塩基の決定精度やコスト面では劣るものの、より断片数が少ない(ギャップの少ない)高品質なゲノム情報が得られる。
リードマッピング解析:メタゲノム解析から得られたゲノムの塩基配列と、ゲノム配列を決定する材料となった元の塩基配列(シーケンサーから出力された配列)を比較する解析。元の塩基配列には「似て非なる」ゲノムに由来する塩基多型や構造多型の情報が含まれる。これらの情報はゲノムを構築する過程で失われてしまうが、リードマッピング解析によって復元することができる。
ショートリードシーケンス:数百塩基のDNA配列断片を低コストで正確かつ大量に解読できるシーケンス技術。メタゲノム解析において現在主流となっている方法である。
門:細菌の分類における最上位の分類階級。以下、綱・目・科・属・種と階級が続く。
古細菌:細菌・真核生物と並ぶ、生物の主要な系統群の1つ。細菌と同様の形・大きさであるが、細胞膜の組成や構造が異なり、細菌とは進化的に異なる生物である。
ゲノム断片数:得られたゲノム配列が、何本のDNA配列断片によって構成されるかを示す。ショートリードシーケンサーによる従来のメタゲノム解析では塩基配列決定長の限界から、1本に繋がったゲノム情報が得られることはまれであり、数百~数千の断片からなるゲノムが得られるのが普通である。
完全長ゲノム:細菌・古細菌のゲノム染色体は1本の環状DNAで構成されるため、完全長ゲノムは1断片の環状配列として得られる。